Cours de chimie Organique -
Les outils du raisonnement en chimie organique
Orbitales moléculaires
Introduction
Théorie des orbitales moléculaires
L'approximation orbitale est basée sur deux hypothèses :
-
la fonction d'onde Y décrivant le
comportement de tous les électrons est un produit de fonctions d'onde
monoélectroniques, appelées orbitales moléculaires (om) ; si l'on
désigne par N, le nombre d'électrons :
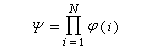
- l'hamiltonien, total du système est la somme d'hamiltoniens monoélectroniques qui n'agissent que sur un seul électron i. Cela revient à négliger les termes d'interaction électronique.
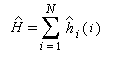
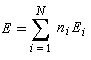
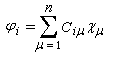
Posons :
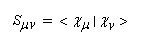
- Hmm s'appelle intégrale coulombienne. Puisque :
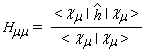
- Hmn est appelée intégrale de résonance (ou intégrale d'échange).
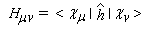
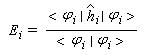
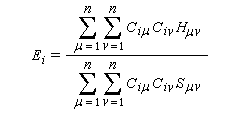
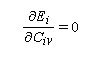
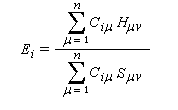
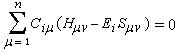
Cette approximation de la théorie des orbitales moléculaires a été introduite par le théoricien allemand E. Hückel en 1931. Elle est connue sous le nom de méthode HMO (Hückel Molecular Orbital). La méthode n'est utilisable en toute rigueur que pour les molécules planes car la notion d'orbitale moléculaire de type p ne possède un sens précis que dans ce cas. D'autre part on s'intéresse uniquement au système des électrons p en partant de l'idée que les orbitales s ont une énergie beaucoup plus basse que celle des orbitales p. On admet alors, en première approximation, que les électrons p se déplacent dans un champ moyen créé par les noyaux et les électrons s du squelette non polarisable de la molécule. Dans ce qui suit on notera cm les orbitales p des atomes de carbone du squelette, qui sont perpendiculaires au plan de la molécule.
Les approximations de Hückel consistent à poser :
Ces approximations sont résumées, avec les notations usuelles, dans le tableau ci-dessous :
|
Hmm
|
Hmn
|
Hmn
|
Smn
|
|
a
|
b (atomes liés)
|
0 (atomes non liés)
|
dmn
|
Il est important de se rappeler que les intégrales a et b sont toutes les deux négatives. Dans le cadre de l'approximation précédente, le système des équations s'écrit :
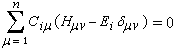
La condition de normalisation se réduit à :
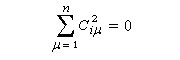
Examinons le cas de l'éthène.
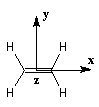
(2) Ci1 + Ci2xi = 0
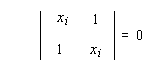
- solution x1 = -1, on a le système suivant :
(1') C11x1 + C12 = 0
(2') C11 + C12x1 = 0
(1') et (2') conduisent l'une et l'autre à :
C11 = C12 - solution x2 = 1, on a un autre système :
(1") C21x2 + C22 = 0
(2") C21 + C22x2 = 0
(1") et (2") conduisent l'une et l'autre à :
C21 = -C22
|
Energie
|
Ci1
|
Ci2
|
|
E1 = a + b
|
0,707
|
0,707
|
|
E2 = a - b
|
0,707
|
-0,707
|
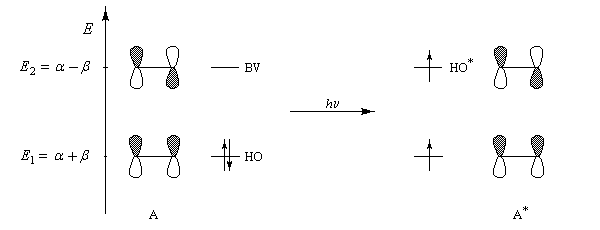
Les orbitales moléculaires p et p* ont respectivement pour expression :
- l'énergie correspondant à l'orbitale moléculaire p (fonction d'onde j1) est plus petite que celle des atomes de départ. Il s'agit d'une orbitale moléculaire liante. On remarque que les orbitales atomiques des atomes interagissent en phase ;
- l'énergie correspondant à l'orbitale moléculaire p* (fonction d'onde j2) est plus grande que celle des atomes de départ. Il s'agit d'une orbitale moléculaire antiliante. On remarque que les orbitales atomiques des atomes interagissent en opposition de phase ;
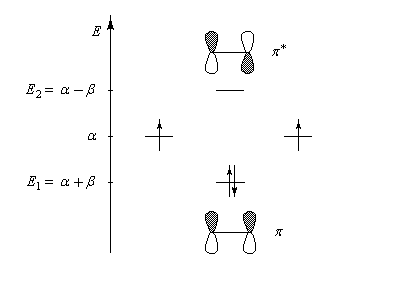
Dans la méthode HMO, les intégrales de recouvrement entre atomes différents sont toutes nulles. Pour caractériser la population de recouvrement, C. A. Coulson a proposé d'introduire la notion d'indice de liaison p :
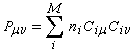
ni est le nombre d'électrons décrit par l'orbitale ji. La somme est étendue aux M orbitales occupées. Dans le cas de l'éthène, on obtient :
La présence d'hétéroatomes, dans le système conjugué est prise en compte grâce à des modèles semi-empiriques.
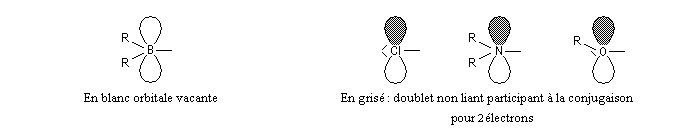
|
|
Nombre d'électrons
|
Intégrale coulombienne
|
Intégrale de résonance
|
|
azote
|
2
|
a N = a + 1,5 b
|
b CN = 0,8 b
|
|
oxygène
|
2
|
a O = a + 2 b
|
b CO = 0,8 b
|
|
chlore
|
2
|
a Cl = a + 2 b
|
b CCl = 0,4 b
|
|
CH3 (modèle hétéroatomique)
|
2
|
a Me = a + 2 b
|
b CMe = 0,7 b
|
Cette méthode a été proposée par R. Hoffman. Elle est basée sur les approximations suivantes :
Le schéma ci-dessous représente ces orbitales moléculaires ainsi que leur énergie relative.
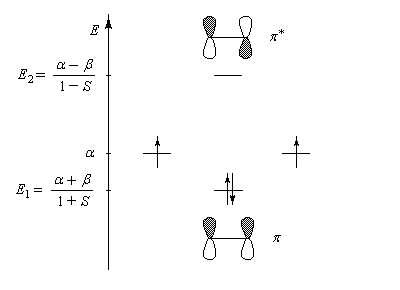
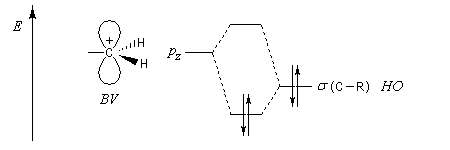
Il est important de noter que l'écart énergétique entre le niveau antiliant et le niveau de départ est plus grand que l'écart entre celui-ci et le niveau liant. La méthode EHMO donne ainsi un résultat plus fidèle à la réalité que la méthode HMO dans laquelle le recouvrement n'est pas pris en considération. Perturbations
Nous nous limiterons aux principaux résultats dans les cas les plus simples. Les calculs sont développées aux références : [21] et [22]
Interaction entre deux om dégénénérées
Interactions entre deux om non dégénérées
Méthode de la résonance
L. Pauling a été le principal promoteur de la théorie de la résonance dans les années 1930. Dans cette méthode, encore appelée méthode de la mésomérie, les noyaux sont supposés fixes (comme dans l'approximation de Born-Oppenheimer). La notion de doublet électronique localisé entre deux atomes subsiste mais cette localisation n'existe que dans certaines formes limites appelée formes limites mésomères. Celles-ci n'ont pas existence réelle. La molécule est un hybride de résonance entre les formes limites, chacune intervenant avec un certain poids statistique.
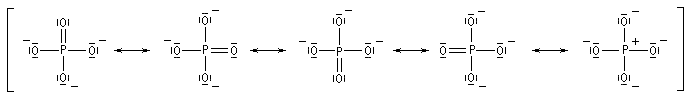
Les différentes formes mésomères ne possèdent pas la même importance dans la description de la molécule réelle. On peut classer ces formes selon leur poids statistique de la manière suivante. Les formes qui contribuent le plus à la structure réelle sont celles qui :
- présentent le plus possible de liaisons covalentes ou ce qui revient au même le moins de séparation de charges ;
- lorsque des charges apparaissent la distance entre elles dans la molécule est la plus grande possible ;
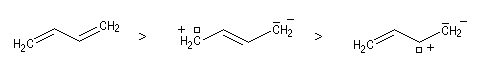
- lorsque des charges apparaissent celles-ci doivent être autant
que possible en conformité avec l'électronégativité de l'atome
correspondant.
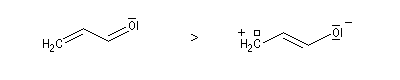
Dans ce qui suit, on désignera par HO, la plus haute orbitale moléculaire occupée (en énergie) de l'un des réactifs et par BV la plus basse orbitale moléculaire vacante de l'autre. En anglais, ces orbitales sont notées respectivement HOMO (highest occupied molecular orbital) et LUMO (lowest unoccupied molecular orbital).
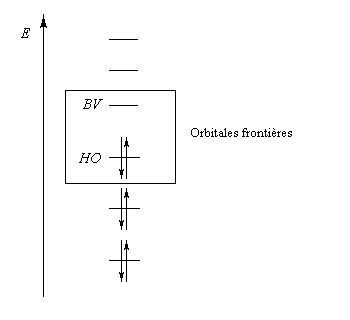
Exemple de la molécule d'éthène
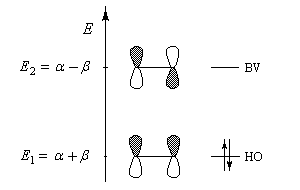 |
Les orbitales frontières de la molécule d'éthène dans le cadre de la méthode de Hückel simple :
|
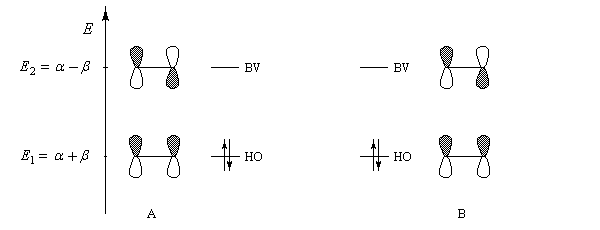
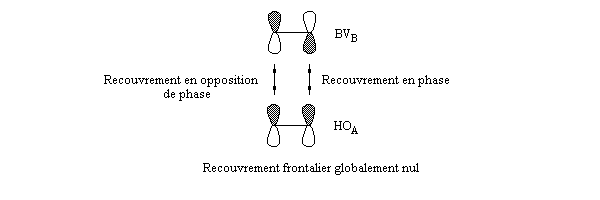
- les interactions entre om remplies sont des interactions à 4 électrons déstabilisantes ;
- les interactions entre om non occupées sont non stabilisantes ;
- les seules interactions stabilisantes (interactions à deux électrons) sont celles entre om remplie d'un réactif et om vide de l'autre réactif.
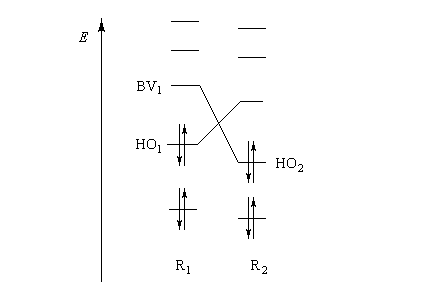
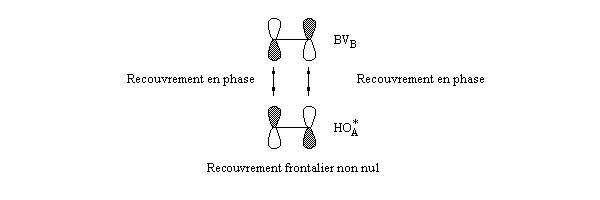
Pourquoi les orbitales frontières ont-elles un tel intérêt ? On peut le comprendre en examinant le diagramme énergétique ci-dessous.
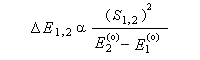
L'interaction dominante est celle qui implique les OF dont l'énergie est la plus proche
Dans l'exemple ci-dessous, il s'agit de l'interaction entre HO1 et BV2 schématisée en trait plein sur la figure.
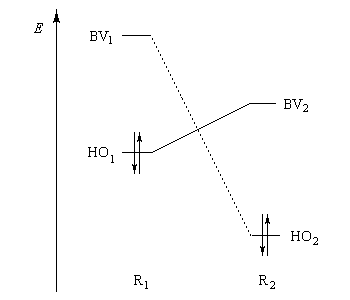
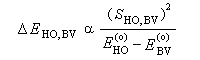
- SHO,BV est plus élevée ; ce fait traduit le principe du recouvrement maximum ;
- la différence d'énergie EHO -EBV est plus petite.
Complément : Equation de Klopman-Salem
Désignons par r et s les atomes qui interagissent lors de l'approche des réactifs R et S avec formation d'un adduit RS. La méthode des perturbations appliquée au système au début de la réaction permet de calculer la différence d'énergie entre RS et R + S. Elle se présente sous la forme de la somme de trois termes :
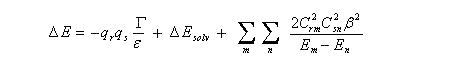
qr, qs : charges des atomes r et s respectivement ;
G : terme de répulsion coulombienne (dépend de la distance entre les charges) ;
e : constante diélectrique du milieu ;
D Esolv : énergie de solvatation ;
Crm, Csn : coefficients pour l'atome r (resp s) dans l'orbitale moléculaire m (resp n) ;
b : intégrale d'échange (intégrale de résonance) décrivant la formation d'une liaison de covalence entre r et s dans l'état de transition ;
Em : énergie de la plus haute orbitale occupée du donneur (HO) ;
En : énergie de la plus basse orbitale vacante de l'accepteur (BV).
Les deux premiers termes traduisent une interaction entre les charges des particules réagissantes tandis que le deuxième terme correspond à l'interaction entre les orbitales frontières du donneur et de l'accepteur. On peut distinguer deux situations limites :
- la différence d'énergie entre les orbitales frontières du donneur et de l'accepteur est grande (En- Em >> 4b 2), les deux premiers termes prédominent. On dit que la réaction est sous contrôle de charge (en anglais : charge controlled).
- la différence d'énergie entre les orbitales frontières du donneur et de l'accepteur est faible Le troisième terme prédomine devant les deux autres. On dit que la réaction est sous contrôle orbitalaire (en anglais : frontier controlled).
Voir : [21] et [22]
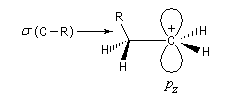
Hyperconjugaison
L'hyperconjugaison consiste en l'interaction entre un nuage électronique de symétrie s et un système de type p ou p.
Examinons à titre d'exemple l'interaction entre des électrons d'une liaison s (C-H) et une orbitale p vide. En termes d'orbitales moléculaires, le phénomène peut être décrit par le diagramme suivant.
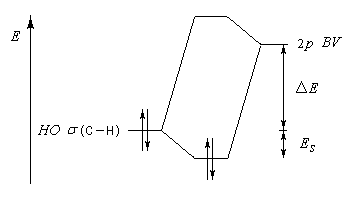
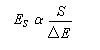
- barrière conformationnelle de l'éthane ;
- stabilisation des ylures ;
- effet anomère.
Electronégativité
L'électronégativité est une grandeur caractérisant l'aptitude d'un atome à conserver ses électrons de valence et à attirer les électrons de ses partenaires. Il existe plusieurs manières de définir l'électronégativité :
- l'échelle de Pauling est basée sur les énergies de dissociation de liaison (grandeurs thermochimiques) ;
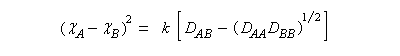
- l'échelle de Mulliken est basée sur l'énergie d'ionisation et l'affinité électronique des atomes.
|
H : 2,1
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Li : 0,98
|
Be : 1,57
|
B : 2,04
|
C : 2,55
|
N : 3,04
|
O : 3,44
|
F : 3,98
|
|
Na : 0,93
|
Mg : 1,31
|
Al : 1,61
|
Si : 1,90
|
P : 2,19
|
S : 2,58
|
Cl : 3,16
|
|
K : 0,82
|
Ca : 1,00
|
Ga : 2,01
|
Ge : 2,01
|
As : 2,18
|
Se : 2,55
|
Br : 2,96
|
|
Rb : 0,82
|
Sr : 0,95
|
In : 1,78
|
Sn : 1,96
|
Sb : 2,05
|
-
|
I : 2,66
|
|
Groupe
|
H
|
CH3
|
C2H5
|
NH2
|
CCl3
|
Ph
|
CN
|
OH
|
NO2
|
|
Electronégativité (Pauling)
|
2,1
|
2,3
|
2,3
|
2,8
|
3,0
|
3,0
|
3,3
|
3,4
|
3,4
|
|
|
tétragonal
|
trigonal
|
digonal
|
|
Electronégativité (Pauling)
|
2,48
|
2,75
|
3,30
|
Le moment dipolaire m d'un composé est une grandeur physique qui peut être mesuré expérimentalement à partir de la permittivité relative de la substance er , et de la densité. Le moment dipolaire m trouve son origine dans l'existence de liaisons partiellement polarisées dans la molécule. Dans la molécule de chlorométhane représentée ci-dessous, la polarisation de la liaison C-Cl s'interprète par la différence d'électronégativité entre les atomes C et Cl. Les liaisons C-H, ne sont pratiquement pas polarisées. La liaison C-Cl se comporte comme un petit dipôle et il est commode de considérer que les atomes C et Cl ont acquis respectivement une charge q = d e et -q = -d e. On représente en général les charges partielles en unité atomique sur les atomes constituant la liaison : d + et d - respectivement. Conformément aux conventions le moment dipolaire est représenté par un vecteur orienté de la charge négative vers la charge positive.
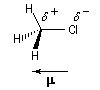
Lorsqu'une molécule possède plusieurs liaisons polarisées, il est commode d'attribuer aux liaisons entre hétéroatomes des moments dipolaires de liaison car l'expérience montre que ces grandeurs sont quasiment caractéristiques des liaisons et sont donc transférables d'une molécule à une autre.
Les moments dipolaires de liaison sont facilement accessibles à partir du moment dipolaire des molécules diatomiques. Pour les molécules polyatomiques, le moment dipolaire d'une molécule est calculé en effectuant la somme des moments dipolaires de liaisons. L'unité usuelle de moment dipolaire est le Debye (D) 1 D = 3,33.10-30 C.m ; bien que n'appartenant pas au système international, cette unité est largement utilisée. Elle a été introduite en souvenir du physico-chimiste néerlandais Peter Debye (prix Nobel de chimie 1936) qui s'illustra dans de nombreux domaines de la physique et de la chimie. Moments dipolaires de quelques liaisons simples (D)
|
Liaison
|
C-H
|
Cl-C
|
I-C
|
N-C
|
O-C
|
F-H
|
I-H
|
|
m (D)
|
0,4
|
1,46
|
1,19
|
0,22
|
0,74
|
1,82
|
0,44
|
|
Composé
|
 |
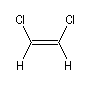 |
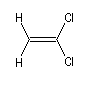 |
|
m (D)
|
0
|
1,3
|
1,9
|
On appelle polarisabilité la propriété physique associée à la création dans une molécule, d'un moment dipolaire induit mi sous l'action du champ extérieur. La polarisabilité est un phénomène dynamique. Le moment dipolaire induit s'annule lorsque la cause qui lui a donné naissance a disparu.
Désignons par E*, le champ ressenti par la molécule dans le milieu où elle se trouve (champ électrique augmenté du champ local de Lorentz [17]).
- elle est d'autant plus élevée que les atomes impliqués sont plus volumineux car les électrons de liaison échappent plus facilement au contrôle exercé par le noyau ;
- elle augmente avec la longueur de la liaison.
|
Composé
|
C-Cl
|
C-Br
|
C-I
|
|
Longueur C-X (pm)
|
178
|
193
|
214
|
|
Moment dipolaire m (D)
|
1,94
|
1,79
|
1,64
|
|
Polarisabilité relative
|
1
|
1,4
|
2,2
|
La polarisation d'une liaison est due à la différence d'électronégativité des atomes liés. Elle se caractérise notamment par l'existence d'un dipôle permanent. Ce dipôle peut induire à son tour une polarisation dans une liaison adjacente polarisable. La transmission de la polarisation à travers des liaisons s par le mécanisme précédent s'appelle "effet inductif".
Les effets inductifs peuvent être classés en deux catégories selon la valeur de leur électronégativité c par rapport à celle de l'atome de carbone (2,5) :
- c > 2,5 effet - I ;
- c < 2,5 effet + I.
|
|
+ I faibles
|
-I forts
|
-I faibles
|
|
-NH2 , -NHR ,
-NR2,
-NHCOR, -O-
|
alkyle
|
-NO2, -CN, -NH3+, -CF3, -N+R3
-CO2H, -CO2R,
-COR -SO3H |
-F, -Cl , -Br , -I, phényle, -OH, -OCH3
|
Lorsqu'on examine la valeur des moments dipolaires descomposés ci-dessous, on constate que le composé aromatique nitré possède un moment dipolaire plus grand que le dérivé saturé correspondant tandis que pour les dérivés fluorés c'est l'inverse.
|
Composé
|
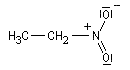 |
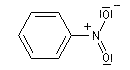 |
|
m (D)
|
3,7
|
4,2
|
|
Composé
|
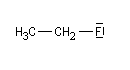 |
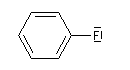 |
|
m (D)
|
1,9
|
1,6
|
Le fluor donne des électrons au cycle. Il s'agit d'un effet donneur.
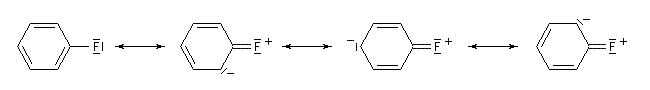
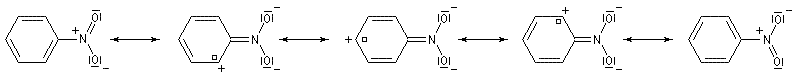
D'une façon générale, lorsque l'effet mésomère et l'effet inductif sont antagoniste, l'effet mésomère l'emporte sur l'effet inductif. Les types fondamentaux de réaction
Définitions
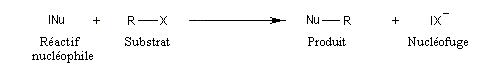
Lorsque deux entités réagissent, il est commode de distinguer le substrat (étymologiquement : sur lequel se déroule le processus) et le réactif. Le substrat est la molécule organique qui "subit l'attaque" du réactif. Lorsque l'une des entités est inorganique, c'est par convention le réactif. Naturellement, il existe une certaine ambiguité lorsque les deux partenaires sont organiques et l'on raisonne alors par analogie. La conversion d'un substrat en un produit particulier indépendamment de la nature des réactifs mis en jeu est une transformation.
La distinction entre substrat et réactif doit être maniée avec précautions mais elle a une certaine importance pour le classement des réactions et de leurs mécanismes. En effet celles-ci sont nommées selon deux critères :
- le type de réaction : addition, substitution élimination ;
- la nature du réactif : électrophile, nucléophile, radicalaire.
|
Réactif
|
électrophile
|
nucléophile
|
|
aspect thermodynamique
|
||
|
aspect cinétique
|
réactivité rapide vis à vis des zones de faible densité électronique
|
réactivité rapide vis à vis des zones de forte densité électronique
|
Classement
La terminologie du classement des réaction est due à Ingold. On distingue neuf types fondamentaux.
|
Additions
|
Substitutions
|
Eliminations
|
|
électrophile AE
nucléophile AN radicalaire AR |
électrophile SE
nucléophile SN radicalaire SR |
électrophile EE
nucléophile EN radicalaire ER |
- addition électrophile des halogénures d'hydrogène sur les composés éthyléniques ;
- halogénation, nitration, par un mécanisme de substitution électrophile en série aromatique.
- élimination nucléophile chez les dérivés halogénés ;
Les carbocations
Introduction
Les carbocations sont des ions positifs du carbone il existe deux familles :
- les plus courants possèdent un sextet d'électrons et sont appelés ions carbéniums ;
- on appelle ions carbonium, les ions du carbone pentacoordiné comme l'ion CH5+.
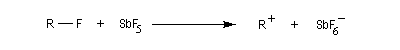
Structure et stabilité des carbocations
La réaction suivante, réalisée en phase gazeuse a été utilisée pour évaluer la stabilité des carbocations. Cela revient à prendre le cation éthyle comme référence des enthalpies libres standard.
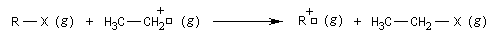
|
Carbocation
|
DrG0 (kJ.mol-1)
|
|
CH3CH2+
|
0
|
|
CH3CH2CH2+
|
-25
|
|
(CH3)2CH+
|
-92
|
|
(CH3)3C+
|
-167
|
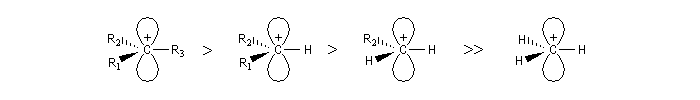
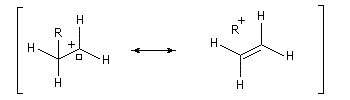
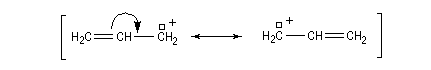
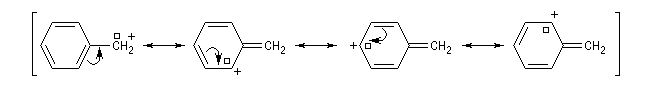
Les ions acylium sont stabilisés par la présence d'un hétéoatome adjacent.
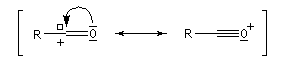
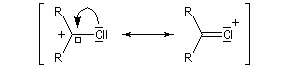
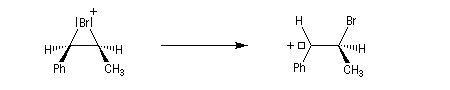
Intervention des carbocations dans les mécanismes
Les carbocations interviennent dans de nombreux mécanismes :
- substitutions nucléophiles monomoléculaires ;
- éliminations nucléophiles monomoléculaires ;
- alkylation de Friedel et Crafts ;
- acylation de Friedel et Crafts ;
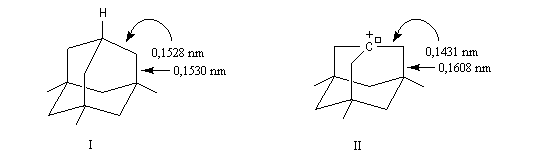
A l'origine, cette transposition a été mise en évidence par Wagner. Il s'agissait de la réaction permettant de passer du chlorhydrate de camphène (I) au chlorure de bornyle (II).
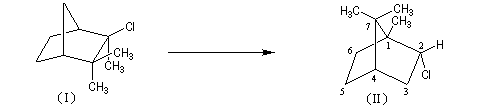
Le mécanisme de la réaction fait intervenir un carbocation qui subit une réaction de transposition.
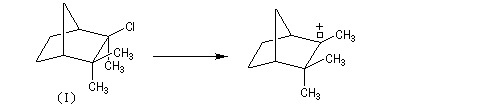

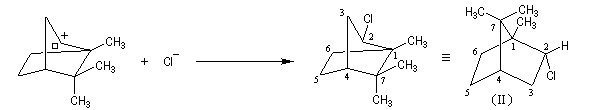
Chaque fois que des carbocations sont générés dans une réaction, on observe ce type de transposition. Exemples :
- déshydratation des d'alcools tertiaires ;
- désamination nitreuse (réaction de Demyanov).
 |
L'adamentane est le tricyclo [3,3,1,1] décane. A la
température ordinaire, ce composé se présente comme un solide cristallin
de couleur blanche, de point de fusion élevé (> 210 °C). Du point de
vue structural, la molécule peut être regardée comme résultant de la
fusion de quatre cycles de type cyclohexane
en conformation chaise. Les angles entre toutes les liaisons sont ceux
qu'on observe dans un tétraèdre régulier d'où l'absence de contrainte
interne dans la molécule. |
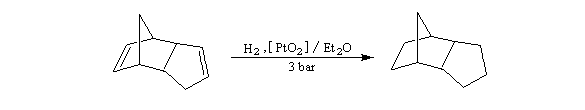
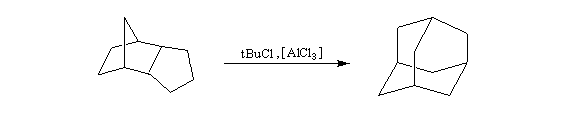

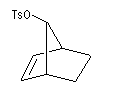
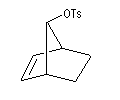
On s'intéresse aux vitesses d'acétolyse des composés suivants.
|
Composé
|
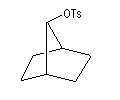 |
 |
 |
|
Vitesse relative
|
1
|
104
|
1011
|
On interprète ces résultats par la formation d'un intermédiaire ponté non classique. Lors de la formation de l'intermédiaire, la double liaison assiste le départ du nucléofuge. On dit qu'il y a eu assistance anchimère du grec ankura : ancre (en anglais : anchimeric assistance ou neighboring group participation).
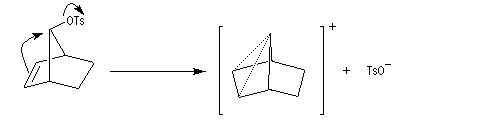
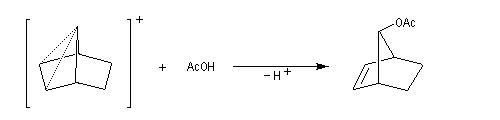
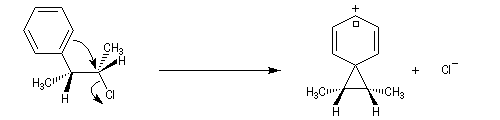
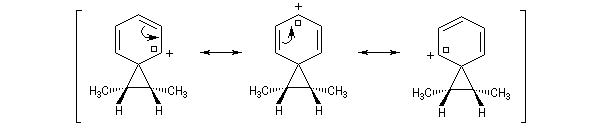
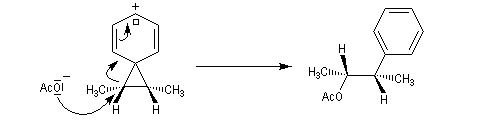
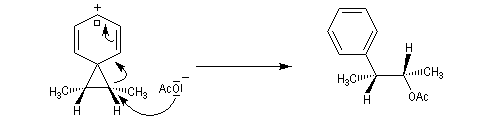
L'étude de la solvolyse des benzènesulfonates d'exo-norbornyle (I) et d'endo-norbornyle (II) a été faite par le chimiste d'origine canadienne Saul Winstein (UCLA) à partir de 1949. Les résultats expérimentaux sont les suivants :
- l'acétolyse de I ou de II conduit exclusivement à des produits de stéréochimie exo ;
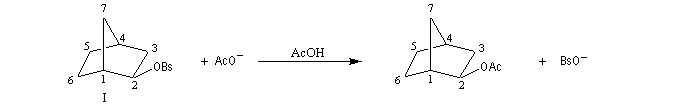
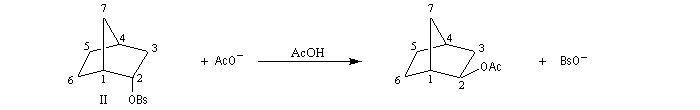
- la vitesse d'acétolyse du benzènesulfonate exo I est 350 fois plus rapide que celle du composé II ;
- l'acétolyse d'un énantiomère de I conduit au mélange racémique du produit final ;
- l'acétolyse d'un énantiomère de II conduit à un mélange non racémique des énantiomères du produit final.
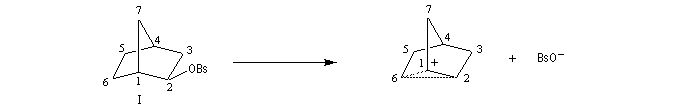
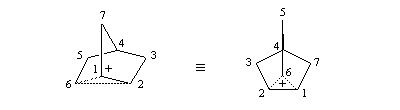
Le cation norbornyle peut être préparé en milieu superacide et son étude par spectroscopie de RMN a été effectuée.
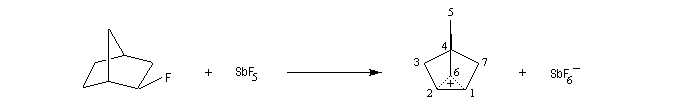
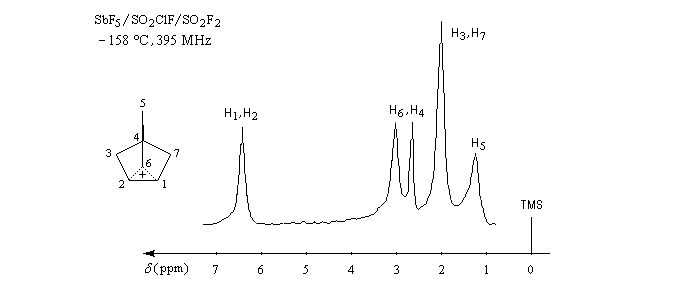
|
Déplacement chimique (ppm)
|
Intensité
|
Attribution
|
|
6,75
|
2 H
|
C1, C2
|
|
3,17
|
2 H
|
C6
|
|
2,82
|
1 H
|
C4
|
|
2,13
|
4 H
|
C3 , C7
|
|
1,37
|
2 H
|
C5
|
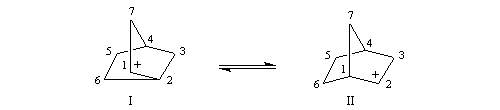
Cette étape est analogue à la réaction de Lucas.
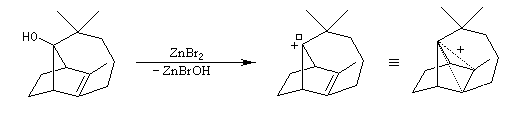
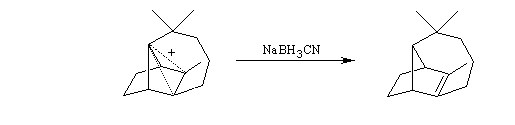
Introduction
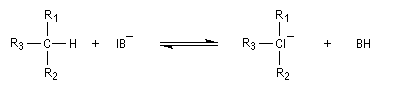

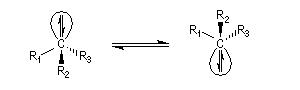
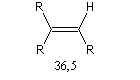
L'acidité croit avec le degré de caractère s.
|
|
tétragonal
|
trigonal
|
digonal
|
|
pKa
|
 |
 |
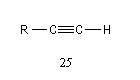 |
A côté des organosodiques proprement dits, les composés dans lesquels la charge négative est stabilisée par délocalisation peuvent être obtenus assez facilement par métallation de la liaison C-H de l'hydrocarbure correspondant avec une base suffisamment forte. Ce sont des sels de sodium dont l'anion est le carbanion formé à partir de l'hydrocarbure.
Un hydrocarbure comme le fluorène (pKa = 23) est suffisamment acide pour pouvoir être déprotoné par l'ion hydroxyde dans le DMSO. On obtient alors un sel de sodium dans lequel l'anion est le carbanion fluorényle.
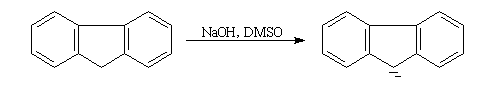
 |
Un mélange d'hydroxyde de sodium en perles et de fluorène sont ajoutés à une petite quantité de DMSO bien sec. Le mélange est agité à la température ambiante. Au bout d'une quinzaine de minutes, on obtient une solution de couleur rouge qui contient l'anion fluorényle. Cet ion est stabilisé par résonance (le cycle central possède un caractère aromatique). La conjugaison est suffisamment étendue pour qu'il absorbe dans la partie visible du spectre. |
- le naphtalène sodium est un intermédiaire de la réduction de Birch. Il est utilisé pour promouvoir des polymérisations anioniques ;
- les radicaux cétyles interviennent dans la réduction des cétones aromatiques.
Parmi les carbanions stabilisés par un groupe fonctionnel les plus importants sont les énolates :
-
énolates de carbonylés
- énolates d'esters
- énolates d'amides
- a-chloroester
- carbanions dérivant de dérivés nitrés.
La présence d'un groupe carbonyle stabilise les carbanions La crotonisation des aldols fait intervenir un carbanion dans un mécanisme de type E1Cb. L'aromaticité est également un facteur de stabilisation. Exemple : l'anion cyclopentadiényle
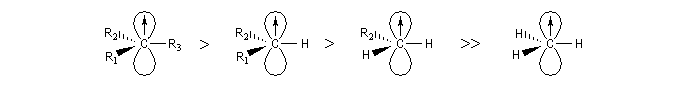
Les radicaux
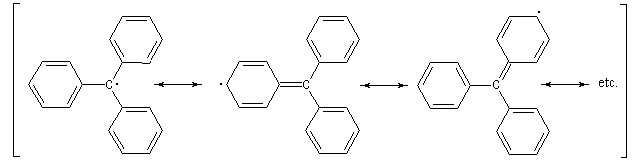
Un radical est une espèce comportant un ou plusieurs électrons non appariés. Le premier radical organique a été mis en évidence par Moses Gomberg en 1900. Il s'agit du radical triphénylméthyle. Le schéma réactionnel utilisé par Gomberg est le suivant :
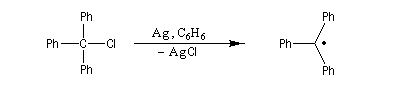
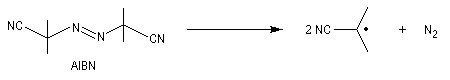
|
Radical
|
Do (kJ.mol-1)
|
|
CH3°
|
435
|
|
CH3CH2°
|
410
|
|
(CH3)2CH°
|
397
|
|
(CH3)3C°
|
385
|
Théorie de l'état de transition
Un complexe activé est une association d'atomes dont l'énergie correspond à un point col sur la surface d'énergie potentielle. L'état de transition thermodynamique est constitué de l'ensemble des complexes activés associés à leurs probabilités de transformation. Notons que beaucoup d'auteurs utilisent sans distinction les termes : complexe activé et état de transition (à l'échelle microscopique dans ce cas). L'état de transition correspond à un état pour lequel le système a une probabilité égale d'évoluer dans le sens direct ou dans le sens inverse.
L'enthalpie libre molaire de référence de l'état de transition est la somme de l'enthalpie libre molaire des réactifs et de l'enthalpie libre d'activation de référence (le symbole typographique utilisé en exposant se lit "double dague".)
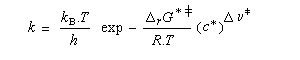

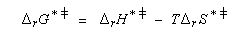
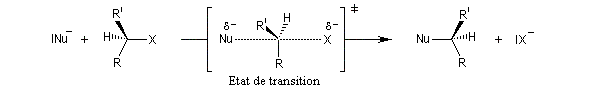
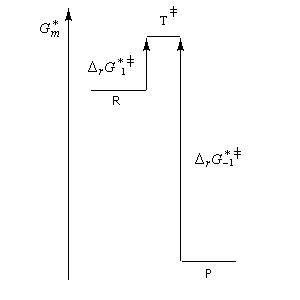
- sur le diagramme I, on a représenté le profil d'enthalpie libre dans le cas où le substrat est un halogénure secondaire ;
- sur le diagramme II, on a fait de même pour un substrat primaire.
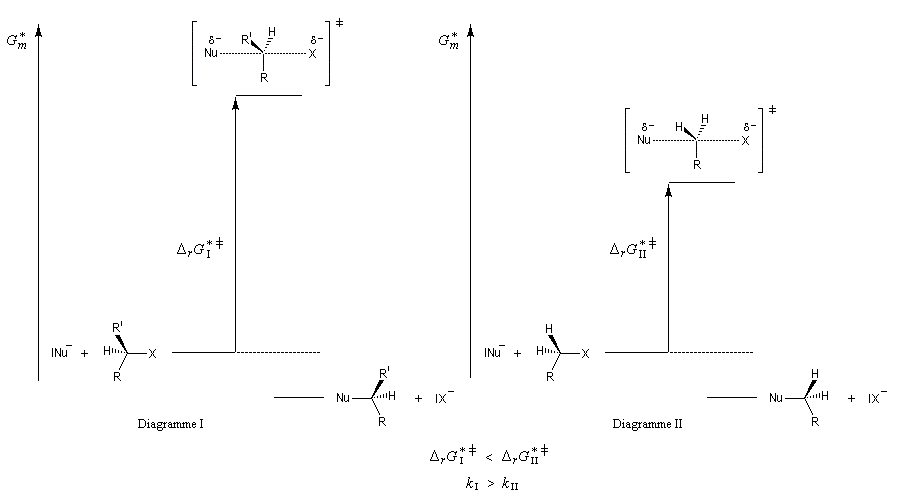
Postulat de Hammond
La connaissance de l'état de transition et de son énergie lors du déroulement d'une réaction sont naturellement d'un grand intérêt notamment lorsqu'il s'agit de l'étape déterminante d'une réaction cinétiquement contrôlée. Malheureusement, un état de transition correspond à une association transitoire d'atomes qui se traduit par un maximum d'énergie. La durée de vie de cette association est tellement courte qu'on ne peut en faire l'étude qu'avec des méthodes très sophistiquées dans des cas peu nombreux. Il est raisonnable d'admettre, dans certains cas du moins, qu'il est possible de ramener l'étude de la structure et de l'énergie d'un état de transition à celle d'une véritable entité chimique pourvu que leurs énergies soient assez proches. C'est l'objet du postulat énoncé par le chimiste américain G. S. Hammond qui peut s'énoncer de la façon suivante :
Une autre façon de formuler la même idée a été formulée par L. E. Leffler quelques années avant Hammond. Elle consiste à dire :
 |
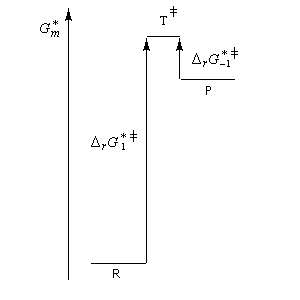 |
| Etat de transition précoce (faible enthalpie libre d'activation). L'état de transition "ressemble" au réactif | Etat de transition tardif (grande enthalpie libre d'activation). L'état de transition "ressemble" au produit. |
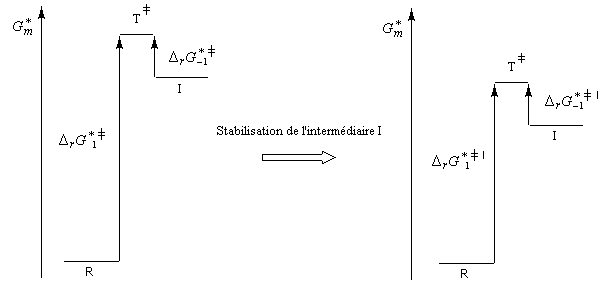 |
| Sous l'influence d'un facteur stabilisant l'intermédiaire I, l'énergie de l'état de transition qui conduit à cet intermédiaire est abaissée. |
Type de contrôle
Définitions
Pour illustrer ces notions nous allons raisonner sur un système particulier. Un réactif A est impliqué dans deux réactions jumelles qui conduisent à des produits notés B et C. On fait l'hypothèse simplificatrice que les réactions directe et inverse sont d'ordre 1. Il s'agit d'étudier l'abondance relative des produits B et C au bout d'une certaine durée de réaction.
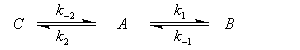
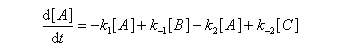
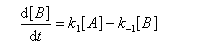
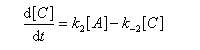
Ce système d'équations peut être simplifié dans deux situations extrêmes.
Contrôle cinétique
Au début de la transformation, les concentrations des produits B et C sont beaucoup plus faibles que la concentration de A. Il est possible de faire les approximations suivantes :
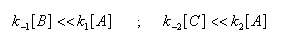
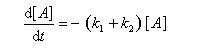
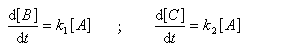
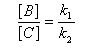
Le rapport des concentrations des produits formés au début de la réaction est égal au rapport des constantes de vitesses. Le produit majoritaire est le produit formé le plus rapidement. On dit que la réaction est sous contrôle cinétique. La proportion des produits formés en cours de réaction fait intervenir la valeur relative des enthalpies libres molaires d'activation.
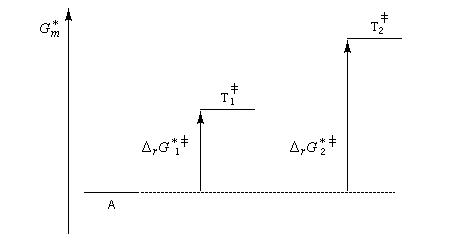
Sous contrôle cinétique, le produit le plus abondant est celui qui se forme le plus rapidement.
Exemples de réactions sous contrôle cinétique :
- addition électrophile des halogénures d'hydrogène sur les composés éthyléniques ;
- halogénation, nitration, par un mécanisme de substitution électrophile en série aromatique.
On s'intéresse désormais à une tranformation renversable dans la fenêtre de durée étudiée, c'est à dire qu'elle peut se dérouler dans le sens direct ou dans le sens inverse. Les vitesses des réactions directe et inverse n'étant pas les mêmes la transformation n'est réellement renversable sur une durée pas trop longue que si ces vitesses sont du même ordre de grandeur.
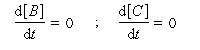
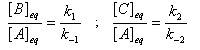
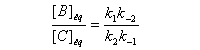
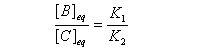
Le rapport des concentrations des produits formés à la fin dela transformation est égal au rapport des constantes thermodynamiques. On dit que la réaction est sous contrôle thermodynamique. Le produit formé majoritairement à l'état final est le plus stable.
Sous contrôle thermodynamique, le produit le plus abondant est celui qui est le plus stable.
Dans le diagramme d'enthalpie libre le produit majoritaire se
caractérise par l'enthalpie libre de réaction de référence la plus
basse.
Exemples de réactions sous contrôle thermodynamique :
- élimination d'eau chez les alcools ;
- sulfonation des composés aromatiques ;
- alkylation de Friedel et Crafts.
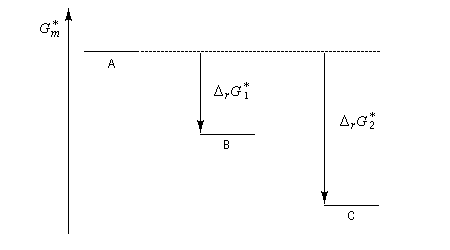
Le type de contrôle peut changer avec la température. Un exemple est celui de la sulfonation du naphtalène :
- à basse température, la réaction est cinétiquement contrôlée. Le produit le plus abondant est celui qui se forme le plus vite. C'est le a-sulfonaphtalène ;
- à température plus élevée, un équilibre entre les produits est possible. Le produit le plus abondant est le plus stable. C'est le b-sulfonaphtalène ;
Définition de Lewis
Un complexe acide-base de Lewis, encore appelé adduit, est le produit de la réaction entre un acide de Lewis et une base de Lewis. Soit une réaction entre un acide de Lewis symbolisé par A et une base de Lewis symbolisée par B conduisant à la formation d'un adduit que nous noterons AB.
- complexes s
- entre un métal de transition et un ligand azoté, par exemple l'ammoniac ou une amine ou un dérivé comme l'EDTA ;
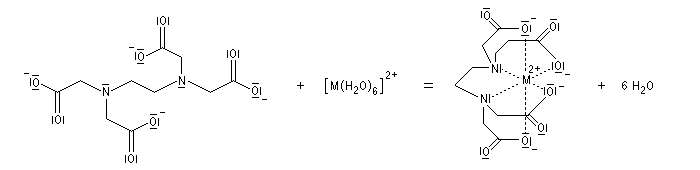
- complexe entre l'éther et BF3, complexe entre l'éther et un organométallique ;
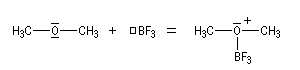
- complexe entre AlCl3 et le composé carbonylé dans l'acylation de Friedel et Crafts ;
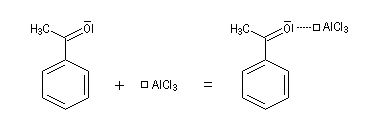
- Complexe entre un ion métallique et un éther couronne ou un cryptand, complexe de Meisenheimer.
- entre un métal de transition et un ligand azoté, par exemple l'ammoniac ou une amine ou un dérivé comme l'EDTA ;
- complexes p
- ferrocène ;
- complexe iode-benzène.
- Acide : toute espèce capable de libérer un proton H+ ;
- Base : toute espèce capable de capter un proton H+ ;
- Couple acide-base : acide et base qui se correspondent par un échange de proton.
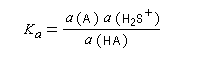
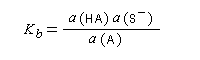
Les méthodes électrochimiques ne permettent de faire des mesures que dans une fenêtre de pKa assez étroite en chimie organique. D'autres méthodes doivent être employées. Le couple menthol-ion mentholate peut être mis à profit pour déterminer les pKa des couples acido-basiques comme alternative aux méthodes électrochimiques car les pouvoirs rotatoires spécifiques de l'alcool et de son sel sont très différents.
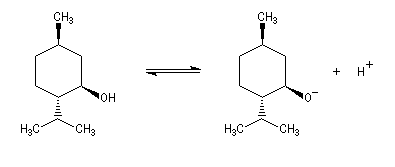
- alcools ;
- phénols ;
- alcynes terminaux
- amines
Les organolithiens sont des bases très fortes permettant de déprotoner les hydrocarbures.
L'acidité cinétique se réfère à la vitesse d'arrachage d'un proton par une base sur un substrat donné. Le tableau suivant, donne pour deux séries d'acides faibles, la constante de vitesse k correspondant à l'arrachement d'un proton par l'eau et la constante d'équilibre Ka de cette réaction.
|
Composé
|
k (s-1)
|
Ka
|
|
CH3COCH3
|
4,7.10-10
|
10-20
|
|
CH3COCH2Cl
|
5,5.10-8
|
3,0.10-17
|
|
CH3COCHCl2
|
7,3.10-7
|
10-15
|
|
CH3CN
|
7,0.10-14
|
10-25
|
|
(CH2)2CN2
|
1,5.10-2
|
10-11
|
L'acide le plus fort pouvant exister dans l'eau est l'ion H3O+ (aq). Tout acide intrinsèquement plus fort protone l'eau pour fournir H3O+ (aq). Dans les années 60, R. J. Gillespie a proposé d'appeler superacides des acides plus forts que l'acide sulfurique pur. Naturellement, une telle définition comporte une part d'arbitraire. Pour aller plus loin, il faut définir une nouvelle échelle d'acidité en milieu non aqueux. L'échelle d'acidité la plus connue est celle de Hammett (Hammett et Deyrup 1930). On utilise une base de référence notée B ci-dessous. Hammett utilisait la nitroaniline. Le rapport des activités de BH+ et B est déterminé par des méthodes spectroscopiques (UV, RMN etc.) Dans cette échelle, l'acide sulfurique concentré est caractérisé par une valeur H0 = - 12.
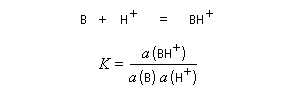
On voit que H0 généralise la notion de pH dans ce type de milieu.
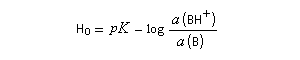
Le mélange équimolaire de HSO3F et de SbF5, découvert dans le laboratoire de G. Olah, a été appelé "acide magique" après qu'un chercheur ait observé la dissolution de la paraffine provenant d'un morceau de bougie tombé par hasard dans le mélange [40]. Il est caractérisé par : H0 = - 19,2. L'acide fluoroantimonique (HF, SbF5) a une valeur H0 = - 31,3.
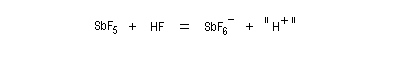
Un tel acide est capable de protoner un hydrocarbure avec formation d'un ion carbonium (I) pentacoordiné puis d'un ion carbénium (II).
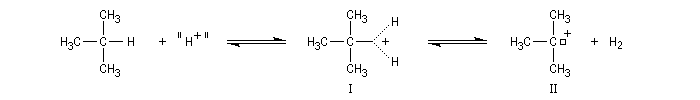
Préparation d'un dérivé du cation norbornyle (Laube 1986)
Les milieux superacides ne sont pas seulement des curiosités de laboratoire.
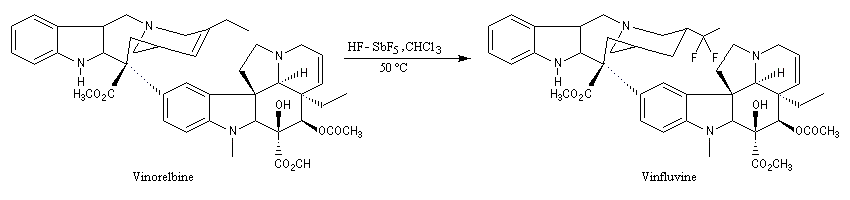
La vinfluvine, un antitumoral, qui fait actuellement l'objet d'essais cliniques, est préparé par fluoration de la vinorelbine en milieu superacide. Le carbocation formé à partir du trichlorométhane arrache un proton du substrat ce qui permet la fixation du fluor.
Une propriété importante d'une base de Lewis est sa nucléophilie. La nucléophilie d'une espèce est une grandeur permettant la comparaison des vitesses relatives de réactions vis à vis d'un même substrat. Il s'agit donc d'une grandeur cinétique relative permettant de classer les nucléophiles selon la vitesse avec laquelle ils transfèrent leur doublet au substrat. La réaction servant de référence est la suivante.
|
Nucléophile
|
pKa (HX/X-)
|
log k/kr
|
Vitesse relative
|
|
CH3OH
|
-3
|
0
|
1
|
|
F-
|
3,4
|
2,7
|
500
|
|
Cl-
|
-5,7
|
4,4
|
2,35´104
|
|
Br-
|
-7,7
|
5,8
|
6,0´105
|
|
I-
|
-5,2
|
7,4
|
2,6´107
|
Lorsqu'on recherche une plus grande force, on peut faire appel aux amidures.
Certains nucléophiles possèdent plusieurs points d'attaque. Ainsi, l'ion nitrite peut conduire par substitution nucléophile sur un dérivé halogéné à des dérivés nitro ou nitrito.
Dureté et mollesse
Principe HSAB de Pearson
Le principe HSAB acronyme de Hard Soft Acid Base principle (en français : principe des acides et bases durs et mous) a été introduit par le chimiste américain G. Pearson en 1963. Afin de rationaliser des faits expérimentaux. Cet auteur distingue trois catégories d'acides et de bases de Lewis : les durs (hard), les mous (soft) et une catégorie intermédiaire (borderline).
On peut l'énoncer de la façon suivante :
- Classification des acides
dursintermédiairesmousH+, Li+, Na+, K+, Mg2+, Al3+, Ce3+, AlCl3, RCO+Fe2+, Co2+, Cu2+, Zn2+, Sn2+, R3C+, SO2Cu+, Ag+, Au+, Hg2+,CH3Hg+ , Pd2+, Pt2+, Br+, I+
Br2, I2, carbènes - Classification des bases
duresintermédiairesmollesOH-, RO- , F- , Cl-, AcO-, RO-
H2O, NH3, ROH, R2O, RNH2ArNH2, C5H5N, N3-, Br-H-, R-, I- , R3P, C6H6, C2H4, CN-, SCN-, R2S, RSH, RS-, SO32-
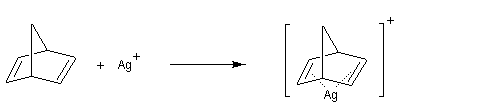
La formation du complexe suivant s'interprète par l'interaction entre le composé éthylénique qui est une base molle et Ag+ qui est un acide mou.
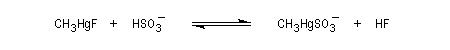
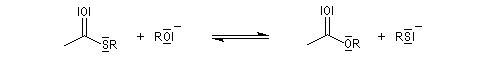
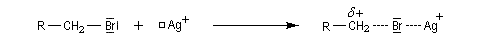
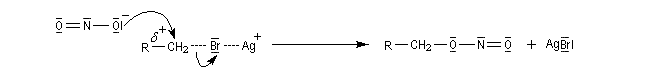
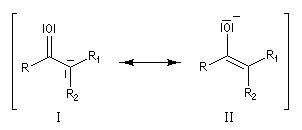
En revanche,avec le chlorure de triméthylsilyle, on obtient une O-alkylation (interaction acide dur-base dure). Cela permet de piéger les énolates sous forme d'éthers d'énols silylés. La mollesse S est définie comme l'inverse de la dureté.
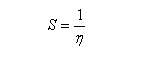
|
molécule
|
dureté h (eV)
|
|
R2CO RSH R2S RNH2 R2NH R3N PhNH2 Py (pyridine) RCN R3P RF RCl RBr RI |
18,8 9,94 9,87 14,7 14,3 14,0 12,1 15,0 15,9 9,42 45,3 10,9 9,54 8,14 |
Considérons un atome d'énergie E comportant N électrons, de charge nucléaire Z. On fait l'hypothèse que E est une fonction continue de N. On envisage une variation dN = N- Z du nombre d'électrons. En effectuant un développement limité de E en fonction de N, on obtient :
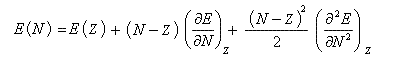
Désignons par I l'énergie d'ionisation et par A l'affinité électronique (opposée de l'énergie de fixation électronique).
On applique les définitions
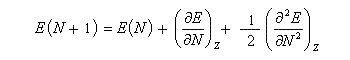
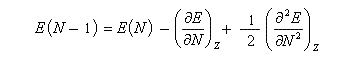
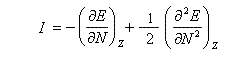
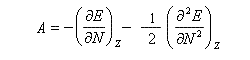
En 1961, R. P. Iczkowsky et J. L. Margrave on proposé de définir l'électronégativité absolue d'une entité (atome, molécule, ion) par la relation :
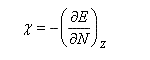
on voit que l'électronégativité s'exprime par la relation ci-dessous :
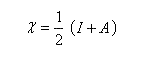
On retrouve une relation qui rappelle la définition de l'électronégativité selon Mulliken mais il faut faire attention que l'électronégativité absolue est une grandeur qui ne caractérise pas en général un seul atome. Il s'agit d'une caractéristique globale de l'entité étudiée :
La dureté est définie par :
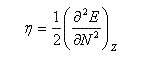
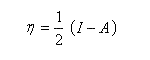
On en déduit une relation entre l'électronégativité absolue et la dureté absolue :
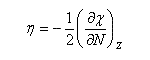
L'expérience montre que la courbe représentant la fonction énergie du système en fonction de N, a l'allure suivante.

Plus précisément, elle peut être assimilée localement à un trinôme du second degré au voisinage de N = Z.
Autrement dit, la courbe qui s'approche le mieux de la courbe expérimentale dessinée plus haut est une portion de parabole. Les coefficients a et b sont des paramètres ajustables qui dépendent du système. Avec cette expression de l'énergie, l'expression de l'électronégativité du système peut être facilement obtenue en dérivant une fois la fonction énergie par rapport à N
Notons que l'électronégativité dépend de N. L'électronégativité du système sans charge excédentaire est la valeur particulière obtenue pour N = Z. C'est une constante.
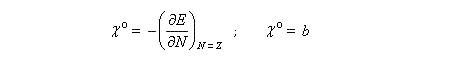
La dureté est obtenue en dérivant deux fois l'énergie. Dans le cadre de ce modèle, c'est une constante.
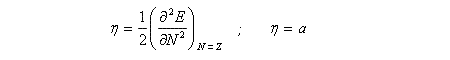
L'électronégativité et la dureté du système sont reliées par la formule suivante.
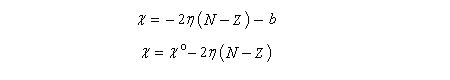
On note l'analogie entre cette dernière expression et celle d'un potentiel chimique. C'est la raison pour laquelle m = - c est appelé potentiel chimique électronique. Avec ces nouvelles notations, l'énergie du système s'écrit :
Application à l'étude de l'interaction entre deux groupes d'atomes avec formation d'une liaison
On s'intéresse à l'interaction entre deux groupes d'atomes A et B avec formation d'une liaison entre A et B dans l'état final. Chaque groupe d'atomes est traité comme un système du type précédent. Tant que l'interaction dure, chaque système est hors d'équilibre. L'évolution va se poursuivre jusqu'à ce que le nouvel ensemble ait atteint un nouvel état d'équilibre.
Le principe d'égalisation des électronégativités énoncé par Sanderson (1951) consiste à admettre que l'équilibre sera réalisé lorsque les électronégativités des deux fragments seront égales dans la nouvelle entité formée. Avant l'interaction, les électronégativités des fragments A et B valent.
Au cours de la transformation, l'une va diminuer tandis que l'autre va augmenter.
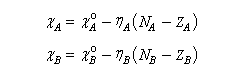
La conservation de la charge permet d'écrire.
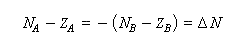
A l'équilibre il y a égalité des électronégativités.
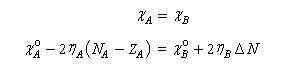
On en déduit :
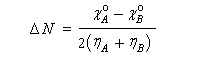
- le sens du transfert de charge est donné par le signe de la différence des électronégativités dans l'état initial. Par exemple si :
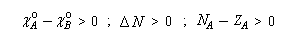
la charge partielle de A va augmenter. - l'intensité du transfert de charge dépend de la différence d'électronégativité et de la somme des duretés. Le transfert entre deux entités peut donc être important même si la différence d'électronégativité n'est pas très élevée entre-elles, à condition que les espèces aient une faible dureté. Ainsi que le fait remarquer Pearson, la dureté apparaît comme une "résistance" au transfert électronique entre les espèces.
L'énergie du sous-système A s'écrit :

de même celle de B
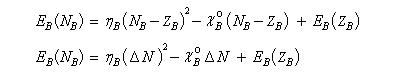
La variation d'énergie du système est la différence :
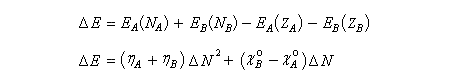
En utilisant la relation établie plus haut :
on obtient finalement :
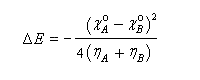
On voit que cette variation d'énergie est négative, ce qui signifie que l'énergie du système a diminué. L'abaissement d'énergie est d'autant plus grand que la différence d'électronégativité est élevée. Mais même si la différence d'électronégativité est faible, l'abaissement d'énergie peut être conséquent s'il implique des espèces de faible dureté. Les résultats précédents peuvent être généralisés dans le cadre d'une théorie beaucoup plus générale : la théorie de la fonctionnelle de densité (DFT). Théorie HSAB et méthode des perturbations
En 1968, Klopman a généralisé et interprété le principe HSAB dans le cadre de l'approximation des orbitales frontières.
Le théorème de Koopmans relie l'énergie des orbitales moléculaires (dans l'approximation de Hartree-Fock) à l'opposé de l'énergie d'ionisation correspondant à l'arrachement de l'électron occupant cette orbitale. Dans le cadre de cette approximation, les énergies des orbitales frontières sont respectivement :
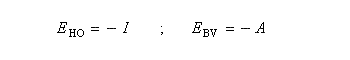
Notons que l'approximation est généralement bonne pour l'énergie d'ionisation mais beaucoup moins pour l'affinité électronique. Si l'on revient aux résultats obtenus plus haut pour l'électronégativité et la dureté :
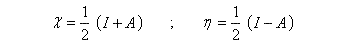
Un calcul très simple permet alors de relier les énergies des orbitales frontières.
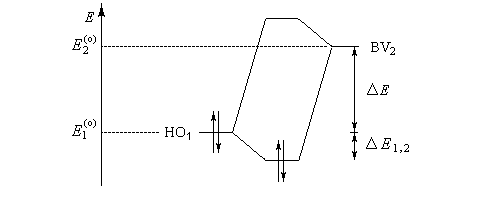
On voit que l'écart énergétique entre HO et BV est directement relié à la dureté.
Le diagramme énergétique ci-dessous résume la discussion précédente.
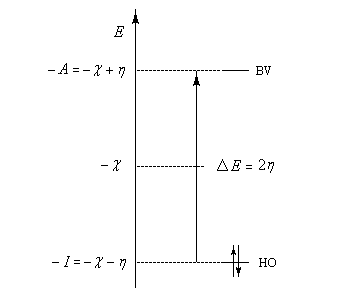
- Une entité dure est caractérisée par une différence d'énergie entre HO et BV élevée :
- une entité molle est caractérisée par une différence d'énergie entre HO et BV faible.
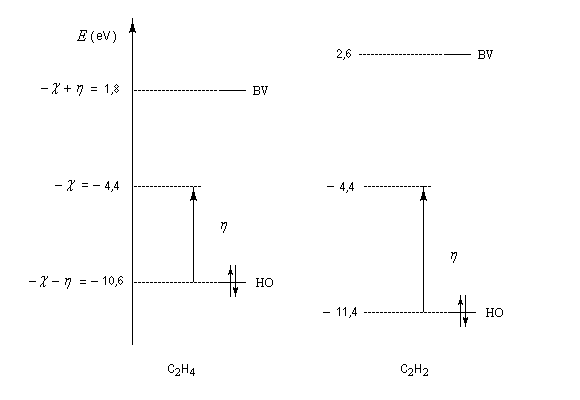
L'électronégativité et la dureté de molécules insaturées simples et de molécules conjuguées peuvent être calculées de façon approchée au moyen de la méthode de Hückel simple.
Dans le cas de l'éthène les énergies des orbitales frontières sont respectivement :
Donc :
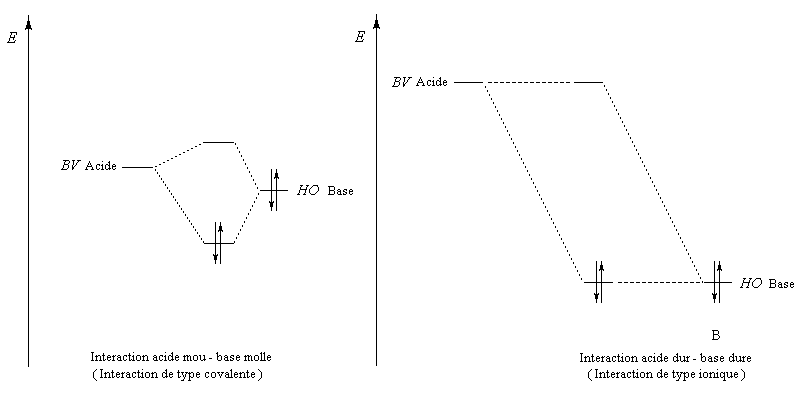
- les interactions sous contrôle orbitalaire correspondent à des interactions entre une base de Lewis et un acide de Lewis pour lesquels la différence d'énergie entre HO et BV est faible. Il s'agit d'intercation entre partenaires mous ;
- le contrôle de charge implique des acides et des bases de Lewis chargés, de petite taille, peu polarisables. Ces espèces recoivent le qualificatif de dures.
|
acides durs (électrophiles)
|
acides mous (électrophiles)
|
|
|
|
bases dures (nucléophiles)
|
bases molles (nucléophiles)
|
|
|
Ouvrages théoriques
[1] F. A. Carey, R. S. Sunberg - Advanced Organic Chemistry, Plenum Press 1990
[2] J. -L. Rivail - Eléments de chimie quantique à l'usage des chimistes, InterEditions du CNRS, 1989.
[3] Atkins, P.W. in Physical Chemistry(Oxford University Press, Oxford, 1986)
[4] P. Atkins - Molecular Quantum Mechanics, Oxford University Press (1983)
[5] N. T. Ahn - Introduction à la chimie moléculaire, Ellipses 1994.
[6] V. Minkine - Théorie de la structure moléculaire, Editions Mir (1979)
[7] Ian Fleming, Frontier Orbitals and Organic Chemical Reactions, J.Wiley & Sons (1976)
Olah G. A., Prakash G. K. S., Sommer J., Superacids, Wiley Interscience, New York, 1985.
Liens
[20] Eléments de chimie quantique
[21] Orbitales frontières, cycloadditions Cours (particulièrement clair) de L. Grimaud, ENSTA.
[22] Introduction à la chimie moléculaire par les orbitales frontières Pascal le Floch, Ecole Poytechnique
Hammond Postulate (goldbook, IUPAC)
Cours d'atomistique du prof. Chaquin, Univ. P et M. Curie
Cours d'atomistique du prof. Silvi, Univ. P et M. Curie
Tableau des pKa des principaux couples acidobasiques (solvant DMSO)
Hyperconjugation by Alan B. Northrup
Gold Book, IUPAC
Equation de Hammett
George A. Olah The Nobel Prize in Chemistry 1994
Cationic rearrangement Prof. Reusch, Michigan State University
Cours de R. Breinbauer.
Adamentane by Paul von R. Schleyer, M. M. Donaldson, R. D. Nicholas, and C. Cupas
HSAB principle hemogenesis, Mark R. Leach
Cours de chimie théorique Caltech Library Service
Utilisation de la méthode des orbitales frontières L. Barriault, Université d'Ottawa
Utilisation de méthodes quantiques semi-empiriques Alain Rochefort, Ecole polytechnique de Montréal
Articles
Hoffmann, R. An Extended Hückel Theory. I. Hydrocarbons. J. Chem. Phys 1963, 39, 1397-1412 (article original : méthode de Hückel étendue)
Fukui K., Yonezawa T., Shingu H., J. Chem. Phys., 1952, 20, 722 (article original de Fukui)
Hammond, G. S. A Correlation of Reaction Rates. J. Am. Chem. Soc. 1955, 77, 334-338. (exposition des bases du postulat de Hammond)
Leffler, J. E. Parameters for the Description of Transition States., Science, 1952, 117, 340-341.
The use and. misuse of the Hammond Postulate, D. Fârcasiu, J. Chem. Educ., 1975, 52, 76.
J. Dellacherie, J.-F. Foucaut et G. Scacchi, l'Actualité chimique, septembre 1980, p. 35-40. (précisions sur la théorie de l'état de transition notamment à propos des états de référence)
J. Dellacherie, J.-F. Foucaut et G. Scacchi, Bulletin de l'Union des Physiciens, juin 1981, N° 635 p. 1235-1245. (version simplifiée de l'article précédent)
W. S. Johnson, J. Am. Chem. Soc., 97, 4777, 1975.
R. G. Pearson, 85, 3533-3543, 1963.(exposition des bases du principe HSAB)
R. G. Pearson, Science, 151, 172-177, 1966. (vulgarisation du concept HSAB)
R. G. Pearson, J. Chem. Ed., 45, 581-587, 1968. (idem)
G. Klopman, J. Am. Chem. Soc., 90, 223-234, 1968. (interprétation du concept HSAB en terme d'orbitales frontières au moyen de la formule de Klopman)
Iczkowsky, R. P ; Margrave, J. L. J. Am. Chem. Soc. 1961, 83, 3547. (une définition de l'électronégativité selon Iczkowsky)
Komorowski, L., J. Chem. Phys., 1987, 114. 55-71. (tableau des duretés d'atomes au sein des molécules)
[40] G. A. Olah, G. K. Surya Prakash, Alain Goeppert, Fluorinated superacidic systems, l'Actualité chimique, octobre-novembre 2006- n° 301-302 (définition et application des milieux superacides)
Robert G. Parr and Ralph G. Pearson (1983). "Absolute hardness: companion parameter to absolute electronegativity". J. Am. Chem. Soc. 105 (26). (définition de la dureté).
J.-P. Foulon, Quelques idées sur la basicité et la nucléophilie, Bulletin de l'Union des Physiciens, novembre 1982, N° 648 p. 143-161.
M. Julia, Intermédiaires réactifs en chimie organique : les carbocations Bulletin de l'Union des Physiciens, mai 1974, N° 648 p. 897-921.
S. Winstein, D. S. Trifan, J. Am. Chem. Soc., 71, 2953 (1949). (travaux sur le cation norbornyle)
R.T. Sanderson, Science, 114,670 (1951) (principe d'égalisation des électronégativités.)
Volkman, Andrews, Johnson, J. Am. Chem. Soc., 97, 4777 (1975).
T. Laube, Angew.Cem. Int. Ed. 1986, 25, 349.










0 commentaires:
Enregistrer un commentaire